MolDesk Basic の主な機能を説明します。
最新版のマニュアルは、以下からダウンロードできます。
- MolDesk Basic クイックマニュアル (Japanese)
- MolDesk Basic マニュアル (Japanese)
- MolDesk Basic Quick Manual (English)
- MolDesk Basic Manual (English)
※ MolDesk Screening は、MolDesk Basic のすべての機能を含みます。
基本機能 [Basic][Screening]
入出力ファイル
| 入力ファイル | mmCIF pdb mol2 sdf mol SMILES (mol2 は multi (多数の構造式を一纏め) も可) (SMILES は 1 行 1 分子の記述で複数分子の入力可能) |
| 出力ファイル | mmCIF pdb mol2 tplファイル (各分子または全系の) エネルギー極小化計算出力 MD計算出力 ドッキング計算出力 (myPresto仕様ファイル) 薬剤スクリーニング結果出力 (CSVファイル MolDesk Screening のみ) |
- mmCIF (PDB) は、4文字のPDB IDを入力してインターネット経由でも入力できます。
- 化合物ファイルは、Compound ID / ID を入力して、PubChem / LigandBox (KEGG DRUG) からインターネット経由で2次元または3次元構造で入力できます。
- オリジナル PDB の残基番号は、結晶水を除いて、計算後も保存しますので、文献の残基番号との照合が容易にできます(保存した例はこちら)。
計算できる分子についての詳細は、こちらをご覧ください。
操作の基本
操作の基本は、マウスによる[選択]、コマンド[実行]、結果の[確認]です。
3D画面やツリー画面で、マウスで選択した対象に対して、操作できるボタンが自動的に表示されます。
(選択できる対象は、原子、残基、チェイン、分子、それらの複数体です。)
シームレスで循環的な実行
操作の履歴は、すべて残っていますので、UNDO、REDOが自由にできます。
MD計算後のドッキング計算、その後にさらに分子を編集してから、MD計算やドッキング計算という、シームレスで循環的な実行が可能です。MolDesk Screening の場合は、Screening 計算も履歴に含めることができます。
プロジェクト作成
一連の編集処理は、プロジェクト単位で保存します。
プロジェクトは、メモリの許す限り、複数作成可能です。一方のプロジェクトが計算中であっても、別のプロジェクトの操作が同時にできます。
プロジェクトの複製、読み込み、エキスポートが可能です。
プロジェクトのエキスポートは、作成した系を、並列計算機やクラウド上で高速計算するときに、入力ファイル一式を持っていくときに使えます。
主な機能 [Basic][Screening]
2D編集と合成容易性をリスト表示
- JChemPaint 最新版により 系の化合物の 2D編集ができます。編集した結果は3次元化して 3D表示します。
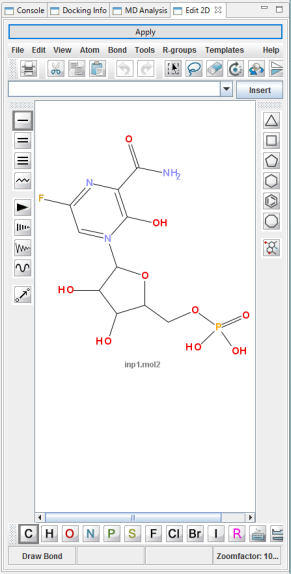
- 系に存在する化合物の 2D構造と合成容易性※ の指標値を、一覧でリスト表示します。
- 分子編集によって分子の構造が変わる度にリアルタイムで表示内容が更新されます。
- 合成容易性は、0(容易)~10(難しい)の指標と、色(緑~黄~赤)で示されます。
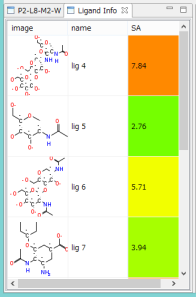
- 化合物の合成容易性を、分子の複雑度、光学活性中心、対称性から予測します。回帰分析なので、10 以上の数値も、1 以下の数字も計算される場合があります。経験的に、10 までなら、なんとか合成できると判断できます。
Prediction of Synthetic Accessibility Based on Commercially Available Compound Databases.
J. Chem. Inf. Model., 2014, 54 (12), pp 3259–3267. (2014)
Yoshifumi Fukunishi, Takashi Kurosawa, Yoshiaki Mikami, and Haruki Nakamura
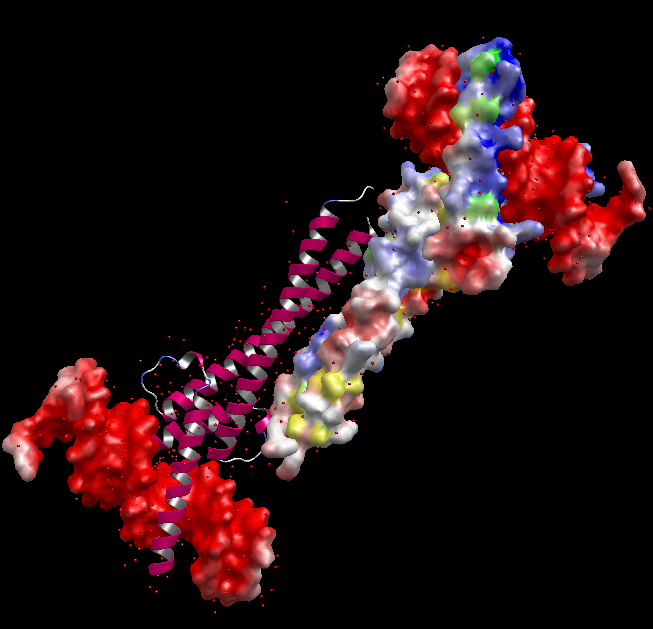
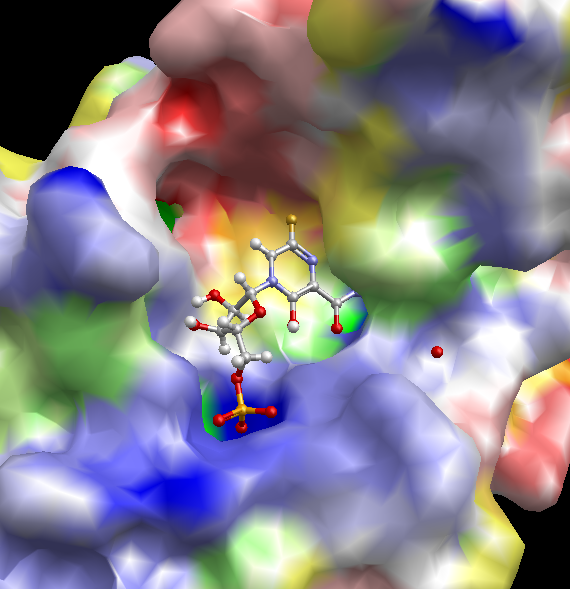
静電ポテンシャル面の表示
- 各 PDB の PDBj eF-site の正確な分子表面(コノリー面、Poisson-Boltzmann 方程式により静電ポテンシャルを計算し色付け)を各分子鎖ごとに表示できます。また、eF-surf / eF-seek で計算した分子表面(ユーザの任意座標構造)も表示できます。
- 青は正、赤は負の電位を表し、黄色~緑色は疎水性残基に対応します。
2D 構造の 3D 構造化
- sdf / mol / mol2 / SMILES ファイルを入力にして、最大、数百万分子の 2D 構造を一括して 3D 構造に変換します。並列処理により高速化してます。
- H 原子の付加は、水素の解離状態は水中での主たるイオン形で行い、AMBER GAFF2 力場で構造最適化し 3次元化して、MOPAC7 AM1、または、AM1-BCC で電荷を付加します。
- 分子1個ごとの Mol2 ファイルと、それらをすべてマージした multi Mol2 ファイルを同時に出力します。
- Conformer(配座異性体)も考慮して変換することが可能です。(4 員環以上の環構造の部分について生成。分子内にキラル中心が存在する場合には、光学異性体も同時に生成。)
- Mol2 ファイルには、下記のように各分子の COMMENT 行に、各種物性値を自動的に付加します。(現バージョンでは、logS, logP は計算できません。)
@<TRIPOS>COMMENT
LIGANDBOX_ID = 00000001-01
SOURCE_ID = NS-00204087
SUPPLIER = ENAMINE
IDNUMBER = Z44490869
MOLECULAR_FORMULA = C13H18N3O
MOLECULAR_WEIGHT = 232.307
MOLECULAR_CHARGE = 1
SUM_OF_ATOMNUMBER = 125
SUM_OF_ATOMNUMBER_MINUS_CHARGE = 124
NUM_OF_DONOR = 3
NUM_OF_ACCEPTOR = 2
HOMO = -12.2444
LUMO = -4.4651
NUM_OF_CHIRAL_ATOMS = 1
NOTE = ENAMINE_Z44490869;Enamine(Fragment)_Z44490869;
タンパク質の40種類のアミノ酸変換
- タンパク質の任意のアミノ酸を、化学修飾した40種類のアミノ酸に変換できます。
- プラス電荷のアミノ酸は、DNAと結合して遺伝情報の読み取りができなくなりますが、中性化すると DNAがほどけて、遺伝情報が読めるようになるなど、アミノ酸の化学修飾は、エピゲノム修飾や、タンパク質を機能させる成熟を考慮する場合に重要です。また、微生物は、20 種類以外のアミノ酸を作るので、微生物にヒトの遺伝子を組み込んで作成したタンパク質は、化学修飾したアミノ酸が重要になります。さらに、コラーゲンはすべて水酸化プロリンです。
- 以下は、20種以外で変換可能な例です。
| 側鎖の H 原子 | ASP (COO–) <–> ASH (COOH) GLU (COO–) <–> GLH (COOH) ARG (C+-NH) <–> ARN (C=N–) LYS (NH3+) <–> LYN (NH2) |
CYS (SH) <–> CYM (S–) CYS (SH) <–> CSO (SOH) PRO (-H) <–> HYP (-OH) |
| OH にリン酸が付加 | SER (OH) <–> SEP (-O-PO3-2) TYR (OH) <–> TYP (-O-PO3-2) |
THR (OH) <–> THP (-O-PO3-2) |
| N にメチル基が付加 | LYS (NH3+) <–> MML (N+H2CH3) LYS (NH3+) <–> DML (N+H(CH3)2) LYS (NH3+) <–> TML (N+(CH3)3) |
ARG (C+-NH) <–> MMA (C+NNCH3) ARG (C+-NH) <–> ADA (C+NN(CH3)2) ARG (C+-NH) <–> SDA (C+(NCH3)2) |
| その他 | SEC (SeH) LYA (Acetyl lysine) NLE (norleucine) HISE, HIS+ |
ポケット作成
- ドッキング計算のプリ処理として、受容体のポケットを表現した、プローブ点の集合を作成します。そのときに、ポケットの中心が必要になります。3D画面またはツリー画面で、マウスで選択してポケットの中心座標を指定します。
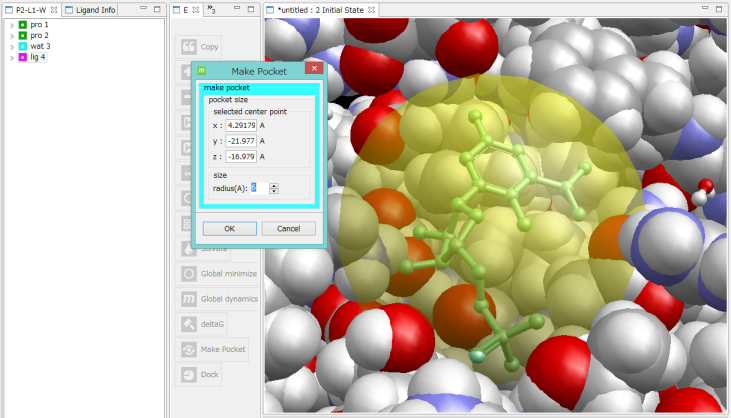
ツリー画面で、lig4分子を選択した例。
このとき、lig4分子の中心座標がポケットの中心になり、ポケットを表す半透明の球を表示します。
ポケットの位置や大きさを変更したいときは、中心座標と半径の値を入力するか、3D画面やツリー画面で原子を複数個、マウスで選択すると、それらの平均座標が中心座標になります。
このため、タンパク質の中心に埋もれたポケットも、容易に選択できます。
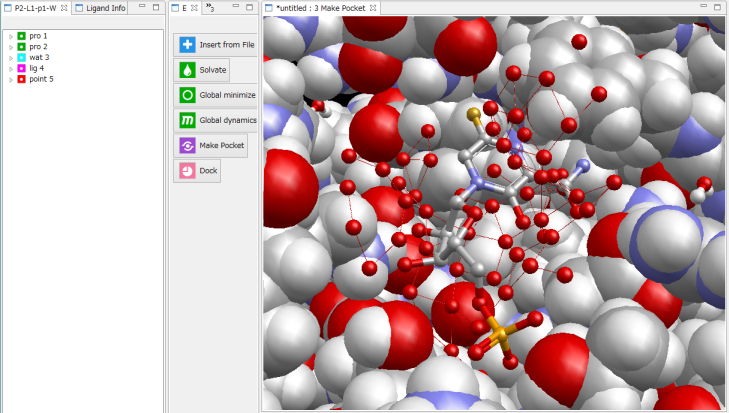
[OK]をクリックして、球内のタンパク質表面にプローブ点を生成した様子。
- 自動的に複数の候補ポケットを作成することもできます。MolDesk Basic では、高速ですが簡易な手法を実装してます。(MolDesk Screening は、1タンパク質あたり約10~20分とやや時間がかかりますが、高精度な手法 (Molsite) を実装してます。)
- ドッキングポーズの正解構造が判明しているリガンド分子の場合は、リガンド分子座標をポケットとして指定できます。
- 上記で作成したポケットや、ポケットを構成するプローブ点で不要ななものは、ユーザがマウス操作で自由に削除可能です。このとき、1分子の編集と同じ扱いになりますので、すべての操作は履歴に残り、編集し直しが可能です。
ポケット探索 (Molsite)
- myPresto の Molecular-docking binding-Site finding (Molsite) 法を利用して、ポケット(蛋白質-リガンド結合サイト)の探索を行います。Molsite によるタンパク質の薬物結合ポケット探索は、計算の初めに、タンパク質の表面に 10Å間隔くらいでポケットを多数設定します。通常、1タンパク質あたり 50個程度のポケットが設定されますが、それぞれのポケットに1万化合物をドッキング計算し、すべてのスコアを評価する手法です。
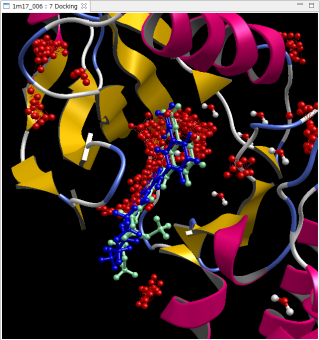
PDB 1m17
青は正解 緑は自動計算で予測したリガンド構造
赤い点は予測したポケット候補
RMSD = 1.36Å
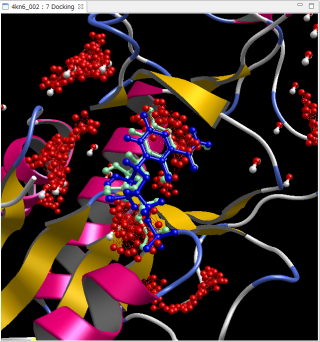
PDB 4kn6
青は正解 緑は自動計算で予測したリガンド構造
赤い点は予測したポケット候補
RMSD = 1.08Å
上図は、PDB 1m17 と PDB 4kn6 で、リドッキング問題を全自動で計算した例です。PDB ファイルを修飾なしでそのまま使用し、[Auto Docking] コマンドで自動的に実行しました。Molsite でポケット探索された複数のポケットの中で、最もスコアが良いポケットに対して自動的にドッキング計算を実行して得られたドッキングポーズです。
- 1タンパク質あたりのポケット探索時間は以下の通りです。この手法も、網羅的なドッキング計算をベースにしているので計算量が大きくスレッド並列計算します。
| 通常のPC Intel Corei7-4790K 4.0GHz 16GBメモリ windows8.1 8並列 |
||
| PDB 1m17 (4744原子) | 15分 | |
| PDB 4kn6 (1555原子) | 11分 | |
Prediction of ligand-binding sites of proteins by molecular docking calculation for a random ligand library.
Protein Science. 20, 95-106. (2011)
Yoshifumi Fukunishi, Haruki Nakamura
ドッキング計算
- ドッキング計算の対象は、1個ないし複数のリガンド分子と、受容体側はタンパク質または核酸分子とその他の分子(水・金属・化合物)の複合体です。
- ユーザは、リガンド分子の多数の sdf / mol / mol2 / SMILES ファイルを一括で指定可で、それらの 3次元化 + 電荷付加 + conformer 生成も一括変換可です (3次元化しない raw ファイルのまま読み込むことも可)。計算系内の1個の化合物を、マウスで選択して指定することもできます。
- ドッキング計算の精度を、precise, moderate, fast から選択します。ドッキングポーズの数を指定します。スコアの高い順に、ドッキングポーズを3D表示します。
- 多数のドッキングポーズをクラスタリングをして、代表構造を3D表示できます。
- タンパク質または核酸分子の複合体が固定で、他のリガンド分子を複数ドッキングさせる場合は、最初に作成したグリッドポテンシャルの使いまわしができます。このとき、数分 —> 数十秒くらいに計算時間を短縮できます。
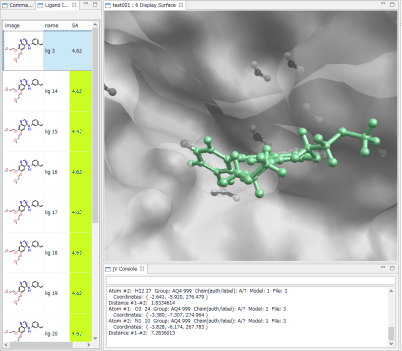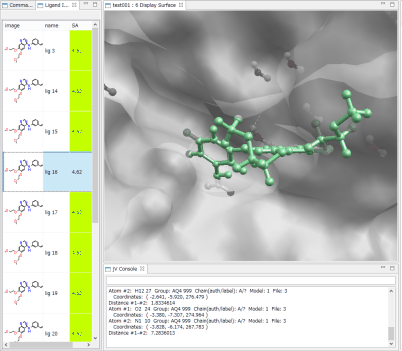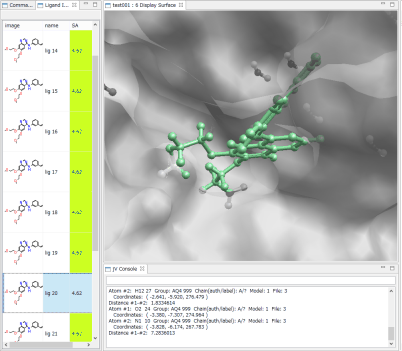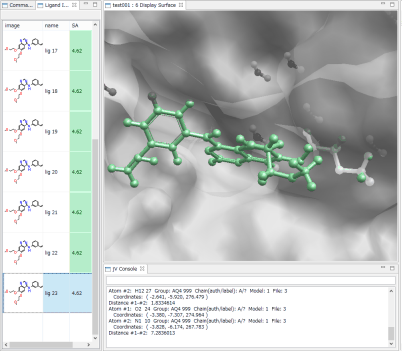
- Ligand Info表示画面で、↑↓キーのクリックだけで、計算されたドッキングポーズ候補の表示の切り替えが、簡単にできます。
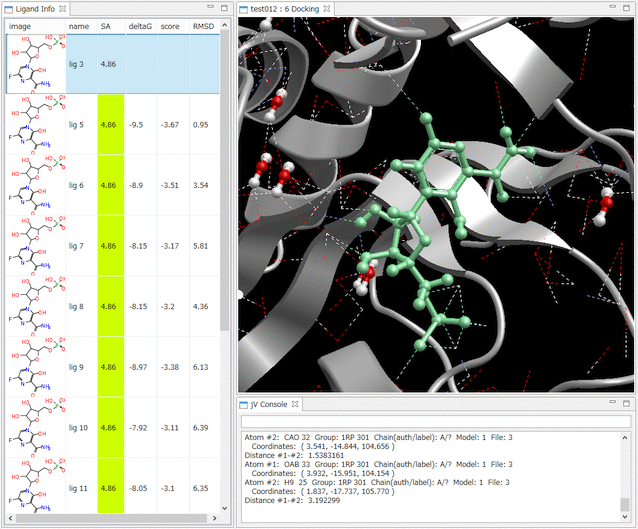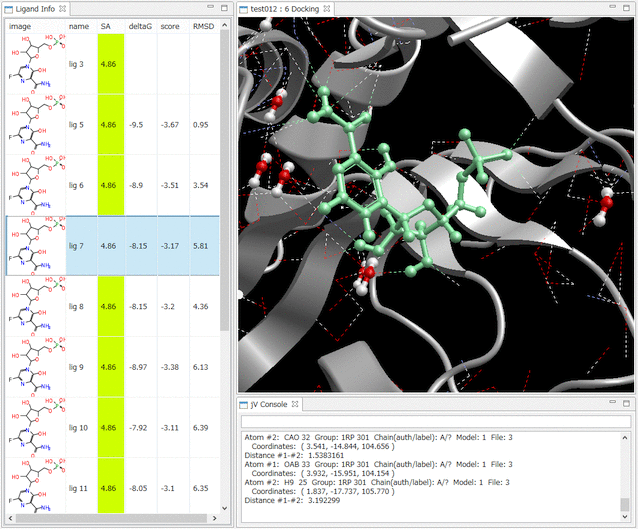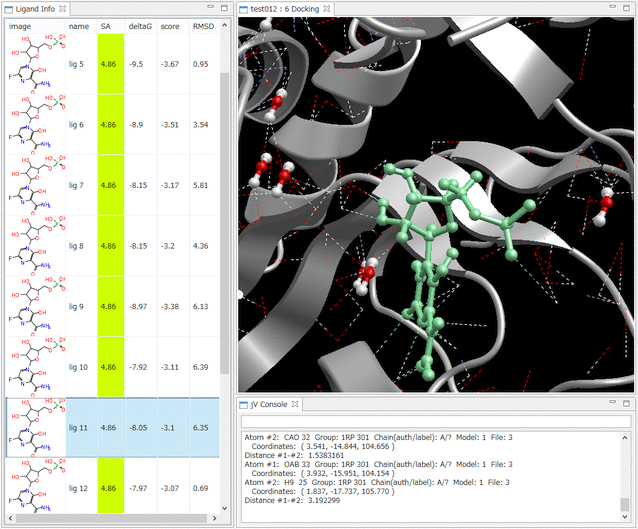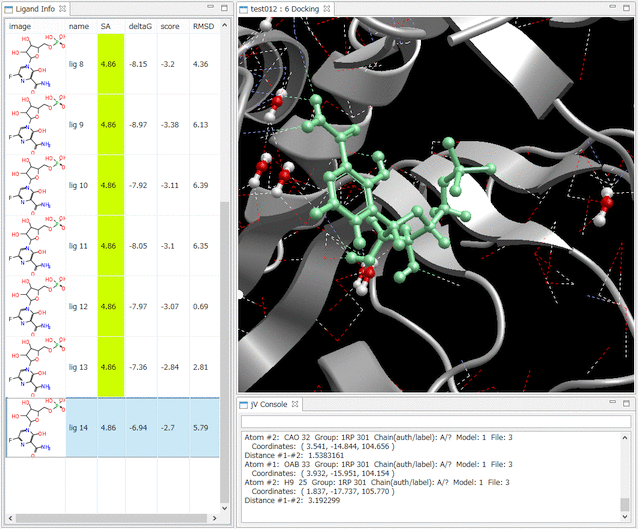
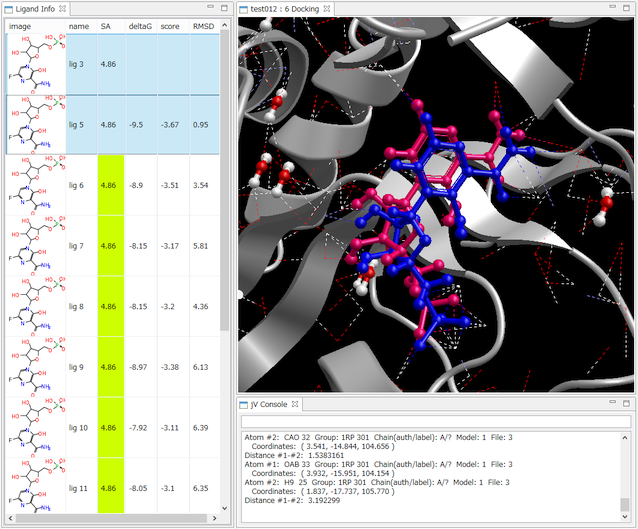
- Ligand Info表示画面で、複数の化合物だけを選択して比較することもできます(下図は、正解構造と最もスコアの良かったドッキング結果の構造の 2つを同時に表示した例)。
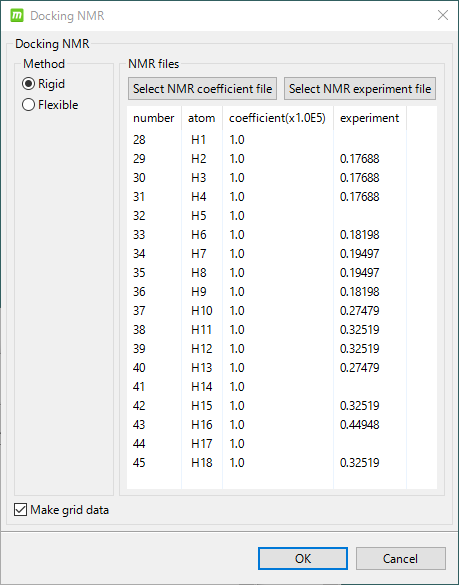
溶液 NMR 実験シグナルによるドッキング補正
- 溶液 NMR の化合物シグナル(水素原子のスピン緩和時間)の実験データ(DIRECTION epitope-mapping)に基づいて、蛋白質-化合物複合体構造を補正することができます。
- ユーザは、水素原子のスピン緩和時間の実験データを、下記画面で入力するか、または、ファイルを入力してから、ドッキング計算をします。下の入力画面例のように、化合物の構造に応じて自動的に入力すべき水素原子を表示します。
Protein-ligand docking guided by ligand pharmacophore-mapping experiment by NMR.
J Mol Graph Model, 31, 20-27, (2011).
Fukunishi Y, Mizukoshi Y, Takeuchi K, Shimada I, Takahashi H, Nakamura H.
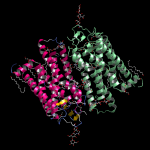
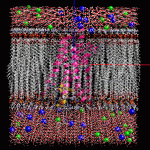
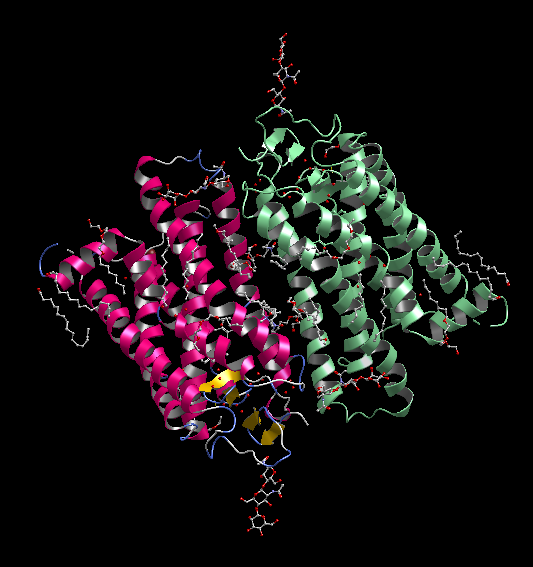
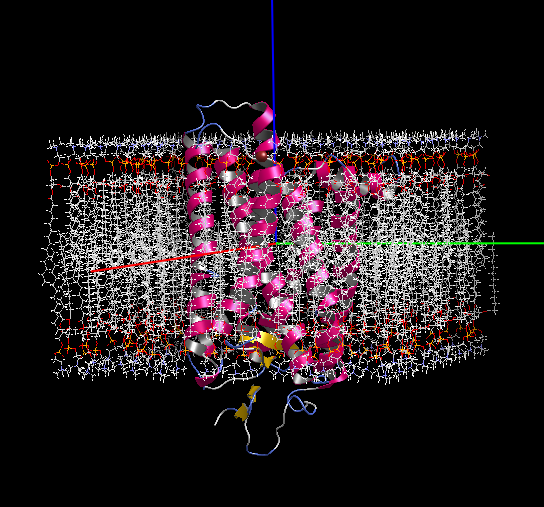
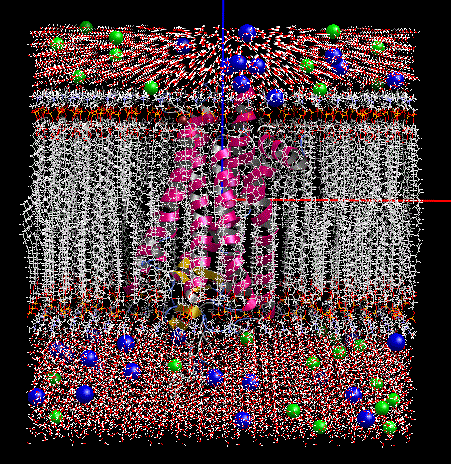
膜貫通タンパク質の脂質2重膜モデリング
-
- 膜貫通タンパク質を入力にして、脂質2重膜中に自動配置します。脂質分子は6種類(DLPC, DMPC, DPPC, POPC, DOPC, POPE)から組成比を設定して選択可能です。イオンで中和した水溶液中に置くこともできます。
- 作成した系は一般にはサイズが大きく原子数も多いので、MolDesk Screening で動作するGPUによる高速 MD プログラム psygene で MD 計算します。脂質分子の力場は AMBER lipid14 を使用します。



{kind=link}
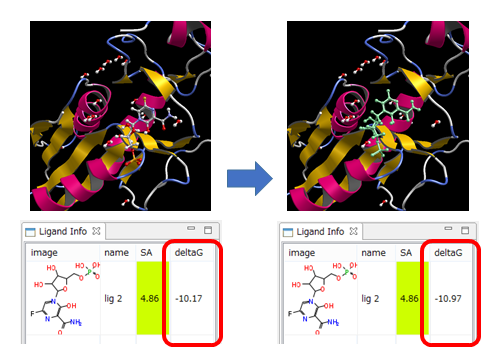
手動ドッキング
- タンパク質のポケット(と推定される所)の近くに、ユーザがリガンドを配置しただけで、ドッキングポーズの構造最適化を行います。
- ポケットの生成は必要ありません。
- 同時に、結合自由エネルギーΔGを計算しますので、ΔG値の減少で、ドッキングポーズ構造の評価ができます。
化合物の構造最適化
- マウスで選択した1個の化合物の、エネルギー最小化計算による構造最適化を実行します。
- マウスで選択したリガンド中の複数の原子を固定して構造最適化できます。 タンパク質のポケット内で、化合物の一部の官能基だけを動かして、それ以外を固定して構造最適化したいとき、などに利用できます。
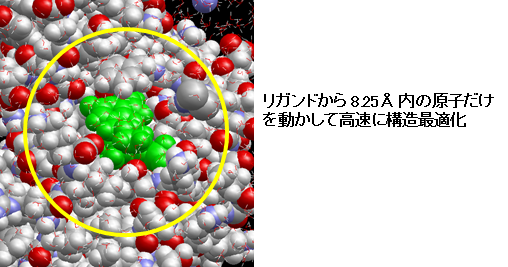
- マウスで選択した1個の化合物と、その周囲 8.25Å の化合物以外の原子だけを構造最適化します。化合物とその周辺のポケット内の原子だけを効率的に最適化計算します。
水分子集団とイオンの付加
Set water
- 溶質のまわりの結晶水の75%を再現する高精度な計算 ( ver.1.1.18 から )。
- 形状が、球または直方体の、水分子の集合を系に付加します。
- 中心座標は、分子団の重心、または、ユーザがマウスで選択した原子で指定できます。
- 球や直方体の大きさの指定方法は、サイズ指定(球の場合は半径、直方体の場合は、XYZ方向長さ)と、分子団の境界からのマージン(Å)の2通りです。
Add ion
Naイオン、KイオンとClイオンを水分子の集合体内に分散します。方法は、
- 自動中和
- イオン濃度(ヒト細胞濃度、ヒト血液濃度、バクテリア濃度、生理食塩水、濃度指定) かつ 自動中和
- イオン数指定
全系の構造最適化
- 使用する力場は、化合物や脂質分子は AMBER GAFF2 など 4種類、その他タンパク質、核酸などは AMBER ff99SB など 5種類の力場から選択できます。( [Build Membrane] で作成した脂質2重膜の脂質分子は、AMBER lipid14 力場を使用します。)
- Steepest decent法、または、Conjugate gradient法で、エネルギー最小化計算による構造最適化を実行します。
- Set waterで設定した系の形状が、球、または、直方体 であるかによって、境界条件の変更を自動的に行います。
- マウスで選択した複数の原子の位置拘束が可能です。または、すべてのタンパク質の主鎖の位置拘束も可能です。
MD計算
- myPresto の MD 計算プログラム ( cosgene, psygene)、または、GROMACS による MD 計算ができます。
- 使用する力場は、タンパク質は、AMBER ff99SB, ff99SB_ILDN_aa, ff99SB_aaなど。RNAは、OL3, LJbb, ROC, Shaw, YILなど。DNAは、OL15など。化合物や脂質分子は AMBER GAFF2 など 。脂質分子は、AMBER lipid14。糖鎖は、GAFF2(GLYCAMには未対応、タンパク質や脂質への結合は保たれません)。
- アンサンブルは、NVT, NPT, NVEから選択できます。系のサイズは、2回目の計算で自動的に引き継ぎます。
- 系の形状が、球、または、直方体 であるかによる、Cap向心力の設定や周期的境界条件の設定は、自動的に行います。球の場合はFMM、直方体の場合はPMEで、クーロン長距離力を計算します。
- リスタート計算時の、上記サイズや、Cap向心力、周期的境界条件の設定も自動設定します(リスタート前の計算条件を引き継ぎます)。
- マウスで選択した複数の原子の位置拘束が可能です。または、すべてのタンパク質の主鎖の位置拘束も可能です。
- Shake計算
MD計算結果(トラジェクトリ)解析
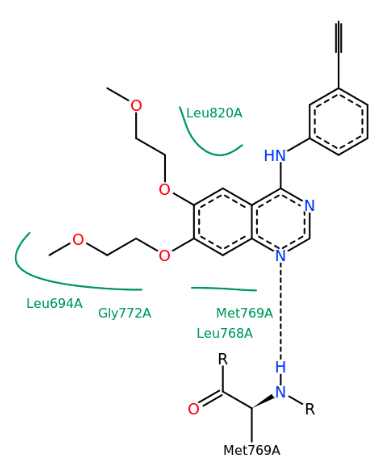
MD 計算のトラジェクトリを動画で確認できます。表示モデルや色の変更など、3D表示で通常行う表示設定を反映します。動画は見たままの形で、アニメーション GIF (animated GIF) ファイルに保存できます。その際、各 Frame 間の時間間隔を変えられます。PDB 1m17 を MD 計算して動画を表示した例。EGFR tyrosine kinase domain の抗がん剤 Erlotinib の分子の、Met769 との水素結合を動画の中で確認できます。
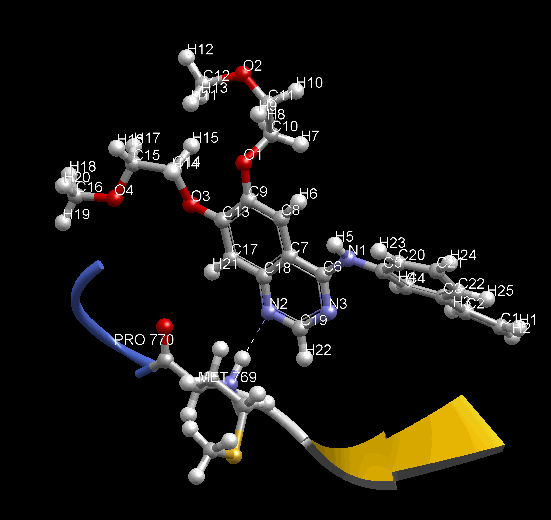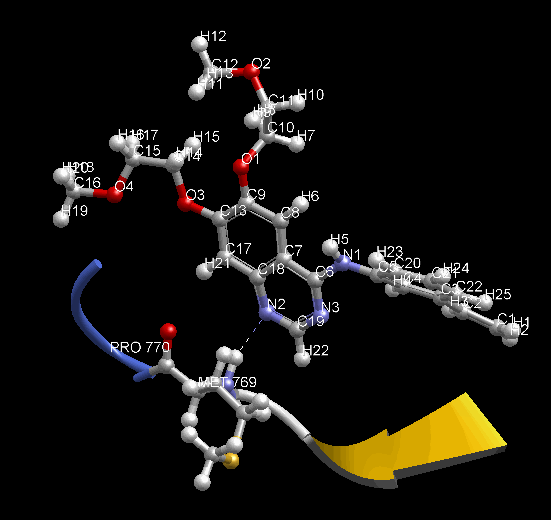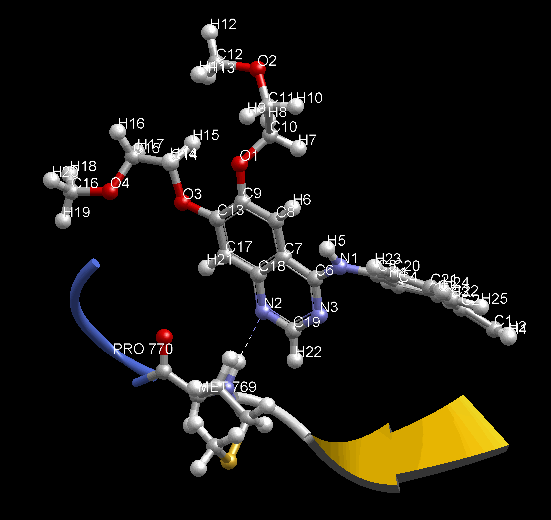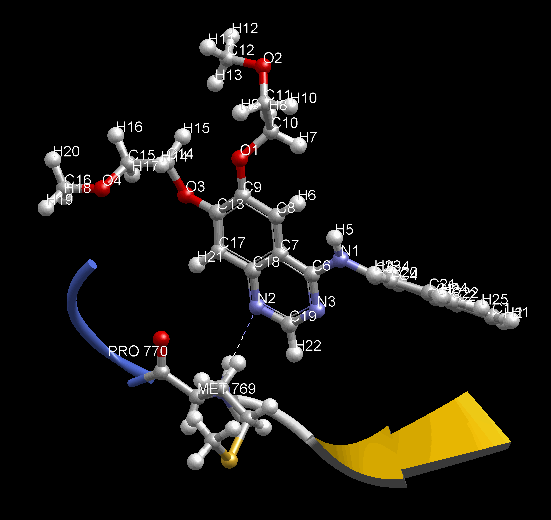
- MD 計算後に、各種エネルギー ( Minimize 計算でも表示可能)、温度、 任意の原子 2 点間の距離、任意の原子 3 点間の角度、任意の原子 4 点の 2 面角、任意の分子の RMSD の時間変化をグラフで確認できます。任意原子は画面上でユーザが選択できます。グラフは動画と時間連動して表示します。
- myPresto 以外の MD プログラムのトラジェクトリファイルと PDB を読み込んで、動画表示できます。対応する MD プログラムは、DCD ( CafeMol / MARBLE / CHARMM / NAMD etc. )、GROMACS、AMBER (netcdf 形式のみ) です。アニメーション GIF (animated GIF) ファイルに保存できます。
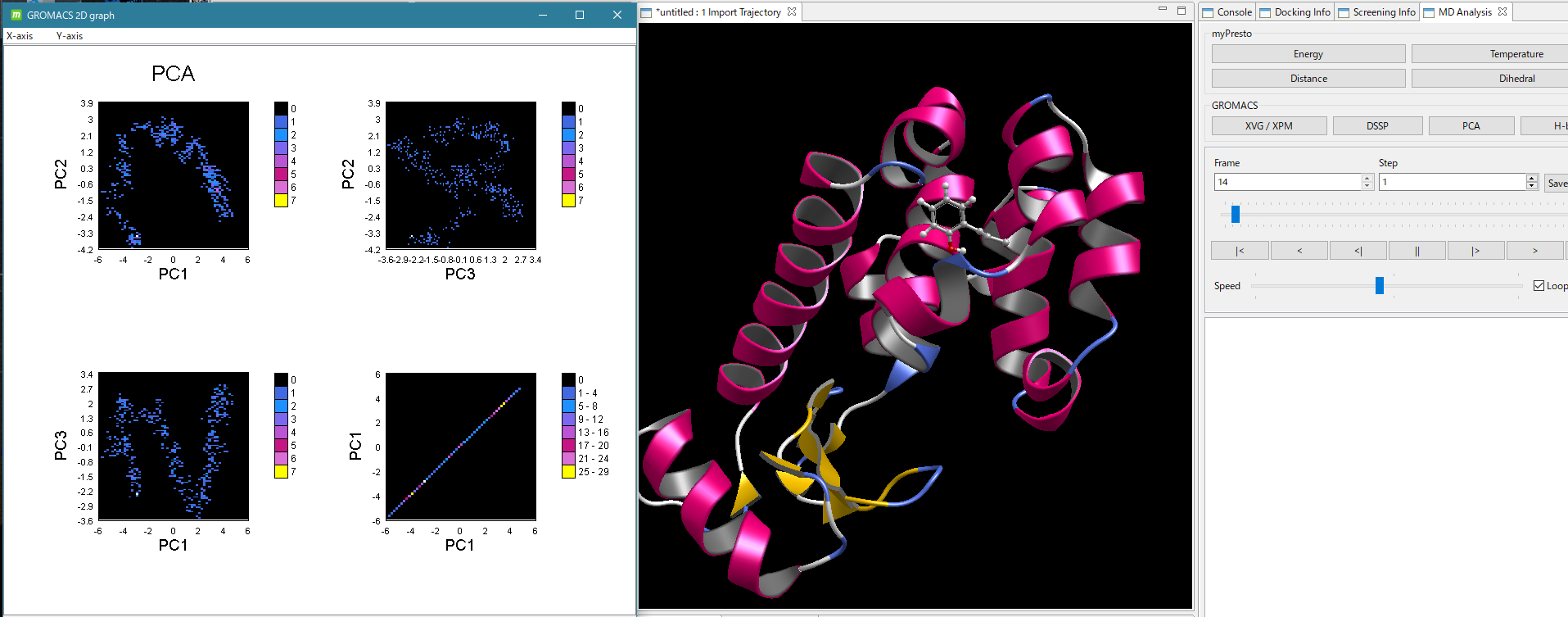
- GROMACS による MD 計算実行後は、GROMACS によるほぼすべてのトラジェクトリ解析が可能です。解析結果の2次元グラフをワンクリックで表示し、動画と時間を同期して表示できます。GROMACSによるMD計算とトラジェクトリ解析の詳しい説明はこちら。
GROMACS で解析した2次構造と水素結合を動画中に時間同期して表示
お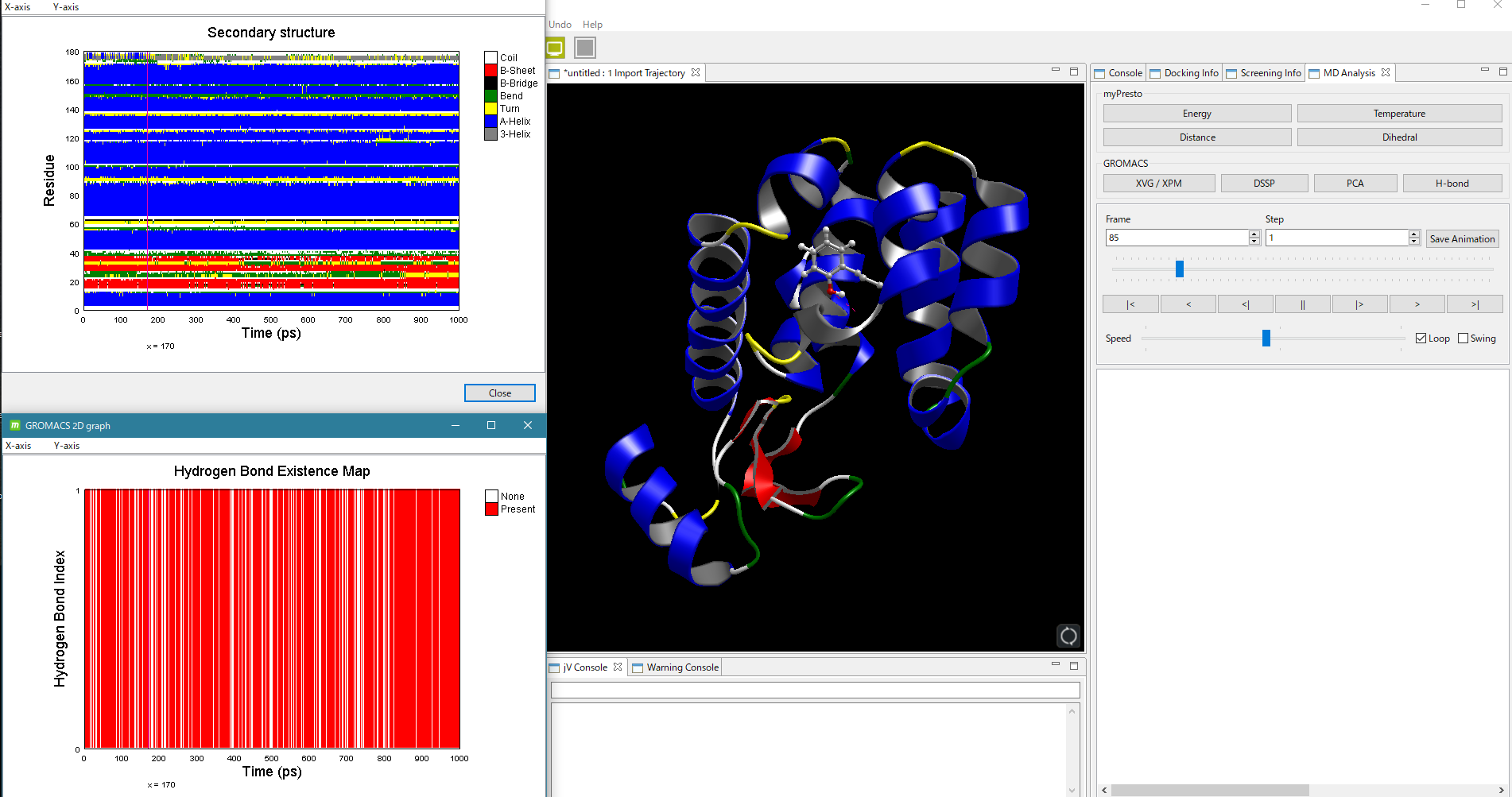 n
n
GROMACS の PCA(主成分)解析を動画と時間同期して表示
(動画の時間に対応した PCA解析グラフの点が白く光る)